Preprocessing the scRNA-seq data#
In this tutorial we will perform some very basic preprocessing steps.
Download data#
The data used for this tutorial is freely available, and can be downloaded from the 10x genomics website.
[87]:
import os
os.chdir("/staging/leuven/stg_00002/lcb/sdewin/PhD/python_modules/scenicplus_development_tutorial/scRNA_seq_pp")
[3]:
!mkdir -p data
!wget -O data/filtered_feature_bc_matrix.tar.gz https://cf.10xgenomics.com/samples/cell-arc/2.0.0/human_brain_3k/human_brain_3k_filtered_feature_bc_matrix.tar.gz
--2024-03-06 13:46:50-- https://cf.10xgenomics.com/samples/cell-arc/2.0.0/human_brain_3k/human_brain_3k_filtered_feature_bc_matrix.tar.gz
Resolving cf.10xgenomics.com (cf.10xgenomics.com)... 2606:4700::6812:ad, 2606:4700::6812:1ad, 104.18.0.173, ...
Connecting to cf.10xgenomics.com (cf.10xgenomics.com)|2606:4700::6812:ad|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 170112442 (162M) [application/x-tar]
Saving to: ‘data/filtered_feature_bc_matrix.tar.gz’
data/filtered_featu 100%[===================>] 162.23M 28.5MB/s in 8.5s
2024-03-06 13:47:00 (19.1 MB/s) - ‘data/filtered_feature_bc_matrix.tar.gz’ saved [170112442/170112442]
[8]:
!cd data; tar -xzf filtered_feature_bc_matrix.tar.gz; cd ..
[45]:
!wget -O data/cell_data.tsv https://raw.githubusercontent.com/aertslab/pycisTopic/polars/data/cell_data_human_cerebellum.tsv
--2024-03-06 14:07:48-- https://raw.githubusercontent.com/aertslab/pycisTopic/polars/data/cell_data_human_cerebellum.tsv
Resolving raw.githubusercontent.com (raw.githubusercontent.com)... 2606:50c0:8001::154, 2606:50c0:8002::154, 2606:50c0:8003::154, ...
Connecting to raw.githubusercontent.com (raw.githubusercontent.com)|2606:50c0:8001::154|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 309204 (302K) [text/plain]
Saving to: ‘data/cell_data.tsv’
data/cell_data.tsv 100%[===================>] 301.96K --.-KB/s in 0.04s
2024-03-06 14:07:48 (7.38 MB/s) - ‘data/cell_data.tsv’ saved [309204/309204]
Preprocessing#
We will do some very basic preprocessing steps, for more information we refer the reader to the Scanpy tutorials.
[9]:
import scanpy as sc
[50]:
adata = sc.read_10x_mtx(
"data/filtered_feature_bc_matrix/",
var_names = "gene_symbols"
)
[51]:
adata.var_names_make_unique()
[52]:
adata
[52]:
AnnData object with n_obs × n_vars = 3233 × 36601
var: 'gene_ids', 'feature_types'
We have already annotated this dataset, this is beyond the scope of this tutorial. We will load this annotation here.
[75]:
import pandas as pd
cell_data = pd.read_table("data/cell_data.tsv", index_col = 0)
cell_data
[75]:
| VSN_cell_type | VSN_leiden_res0.3 | VSN_leiden_res0.6 | VSN_leiden_res0.9 | VSN_leiden_res1.2 | VSN_sample_id | Seurat_leiden_res0.6 | Seurat_leiden_res1.2 | Seurat_cell_type | |
|---|---|---|---|---|---|---|---|---|---|
| AAACAGCCATTATGCG-1-10x_multiome_brain | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_1 (1) | MOL_B_3 (6) | 10x_multiome_brain | NFOL (1) | MOL (1) | MOL |
| AAACCAACATAGACCC-1-10x_multiome_brain | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_3 (5) | MOL_B_4 (4) | 10x_multiome_brain | NFOL (1) | NFOL (3) | NFOL |
| AAACCGAAGATGCCTG-1-10x_multiome_brain | INH_VIP | INH_VIP (6) | INH_VIP (8) | INH_VIP (8) | INH_VIP (10) | 10x_multiome_brain | INH_VIP (7) | INH_VIP (6) | INH_VIP |
| AAACCGAAGTTAGCTA-1-10x_multiome_brain | MOL_A | MOL_A (1) | MOL_A_2 (1) | MOL_A_1 (0) | MOL_A_2 (0) | 10x_multiome_brain | NFOL (1) | NFOL (3) | NFOL |
| AAACCGCGTTAGCCAA-1-10x_multiome_brain | MGL | MGL (7) | MGL (10) | MGL (10) | MGL (12) | 10x_multiome_brain | MGL (8) | MGL (9) | MGL |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| TTTGTGAAGGGTGAGT-1-10x_multiome_brain | INH_VIP | INH_VIP (6) | INH_VIP (8) | INH_VIP (8) | INH_VIP (10) | 10x_multiome_brain | INH_SST (5) | INH_SST (8) | INH_SST |
| TTTGTGAAGTCAGGCC-1-10x_multiome_brain | AST_CER | AST_CER (2) | AST_CER (2) | AST_CER (2) | AST_CER_1 (7) | 10x_multiome_brain | BG (2) | BG (2) | BG |
| TTTGTGGCATGCTTAG-1-10x_multiome_brain | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_1 (1) | MOL_B_1 (1) | 10x_multiome_brain | MOL (0) | MOL (1) | MOL |
| TTTGTTGGTGATCAGC-1-10x_multiome_brain | MOL_A | MOL_A (1) | MOL_A_2 (1) | MOL_A_1 (0) | MOL_A_1 (11) | 10x_multiome_brain | NFOL (1) | NFOL (3) | NFOL |
| TTTGTTGGTGATTTGG-1-10x_multiome_brain | INH_SST | INH_SST (5) | INH_SST (7) | INH_SST (7) | INH_SST (9) | 10x_multiome_brain | INH_SST (5) | INH_SST (8) | INH_SST |
2392 rows × 9 columns
We modify the index of this cell type annotation dataframe so that the cell barcode names match with those in the AnnData object.
[76]:
cell_data.index = [cb.rsplit("-", 1)[0] for cb in cell_data.index]
[64]:
adata = adata[list(set(adata.obs_names) & set(cell_data.index))].copy()
[65]:
adata.obs = cell_data.loc[adata.obs_names]
[81]:
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
Data normalization#
It’s important to save the non normalized and non scaled matrix in the raw slot!
[66]:
adata.raw = adata
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata = adata[:, adata.var.highly_variable]
sc.pp.scale(adata, max_value=10)
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/scenicplus_development_tutorial/lib/python3.11/site-packages/scanpy/preprocessing/_simple.py:843: UserWarning: Revieved a view of an AnnData. Making a copy.
view_to_actual(adata)
[69]:
adata.obs
[69]:
| VSN_cell_type | VSN_leiden_res0.3 | VSN_leiden_res0.6 | VSN_leiden_res0.9 | VSN_leiden_res1.2 | VSN_sample_id | Seurat_leiden_res0.6 | Seurat_leiden_res1.2 | Seurat_cell_type | |
|---|---|---|---|---|---|---|---|---|---|
| ACCTGTTGTGGATTCA-1 | MOL_A | MOL_A (1) | MOL_A_2 (1) | MOL_A_1 (0) | MOL_A_2 (0) | 10x_multiome_brain | MOL (0) | MOL (0) | MOL |
| CTAAAGCTCCCGCCTA-1 | INH_VIP | INH_VIP (6) | INH_VIP (8) | INH_VIP (8) | INH_VIP (10) | 10x_multiome_brain | INH_VIP (7) | INH_VIP (6) | INH_VIP |
| AAGGAAGCACATAACT-1 | AST_CER | AST_CER (2) | AST_CER (2) | AST_CER (2) | AST_CER_2 (5) | 10x_multiome_brain | BG (2) | BG (2) | BG |
| GAGCATGCAATATGGA-1 | MOL_A | MOL_A (1) | MOL_A_2 (1) | MOL_A_1 (0) | MOL_A_2 (0) | 10x_multiome_brain | MOL (0) | MOL (0) | MOL |
| GCTCACAAGACAAGTG-1 | MOL_A | MOL_A (1) | MOL_A_2 (1) | MOL_A_1 (0) | MOL_A_2 (0) | 10x_multiome_brain | MOL (0) | MOL (1) | MOL |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ACATTGCAGCGGATTT-1 | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_1 (1) | MOL_B_3 (6) | 10x_multiome_brain | NFOL (1) | NFOL (3) | NFOL |
| ACGCCTTTCGACCTGA-1 | AST | AST+ENDO (9) | AST+ENDO (6) | AST+ENDO (13) | AST (15) | 10x_multiome_brain | AST+ENDO (6) | AST (7) | AST |
| CTCAGGATCCACCTGT-1 | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_1 (1) | MOL_B_1 (1) | 10x_multiome_brain | NFOL (1) | MOL (1) | MOL |
| CCAACATAGATAACCC-1 | MOL_A | MOL_A (1) | MOL_A_1 (9) | MOL_A_1 (9) | MOL_A_1 (11) | 10x_multiome_brain | NFOL (1) | COP (10) | COP |
| ATGTAAGCACTTAGGC-1 | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_1 (1) | MOL_B_1 (1) | 10x_multiome_brain | MOL (0) | MOL (0) | MOL |
2313 rows × 9 columns
[70]:
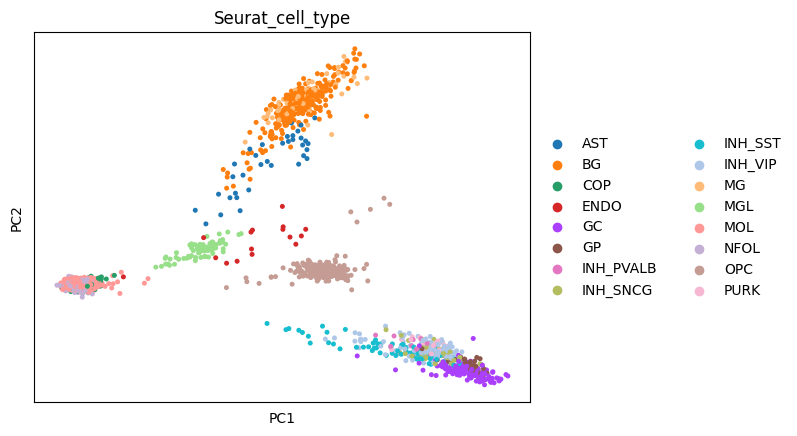
sc.tl.pca(adata)
sc.pl.pca(adata, color = "Seurat_cell_type")
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/scenicplus_development_tutorial/lib/python3.11/site-packages/scanpy/plotting/_tools/scatterplots.py:378: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
[77]:
sc.pp.neighbors(adata)
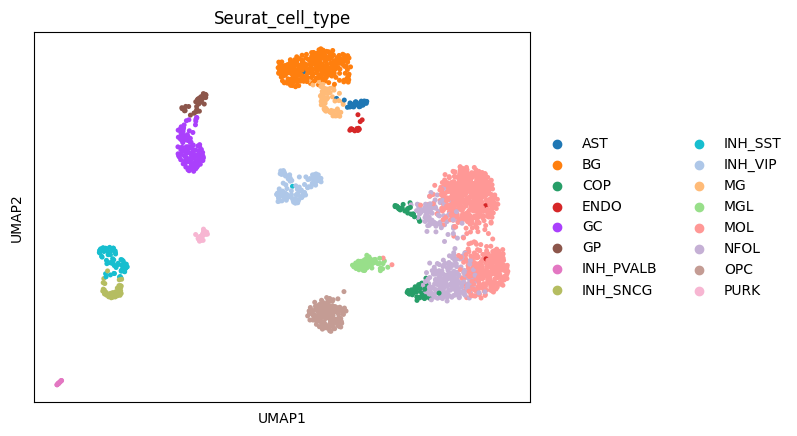
[78]:
sc.tl.umap(adata)
[84]:
sc.pl.umap(adata, color = "Seurat_cell_type")
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/scenicplus_development_tutorial/lib/python3.11/site-packages/scanpy/plotting/_tools/scatterplots.py:378: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
[88]:
adata.write("adata.h5ad")