API#
SCENIC+ object#
Combine single-cell expression data and single-cell accessibility into a single SCENIC+ object.
This object will be used for downstream analysis, including: region-to-gene and TF-to-gene linking, enhancer-driven-GRN building and further downstream analysis.
This object can be generated from both single-cell multi-omics data (i.e. gene expression and chromatin accessibility from the same cell), and seperate single-cell chromatin accessbility data and single-cell gene expression data from the same or similar sample.
In the second case both data modalities should have a common cell metadata field with values linking both modalities (e.g. common celltype annotation).
- class scenicplus.scenicplus_class.SCENICPLUS(X_ACC: spmatrix | ndarray | DataFrame, X_EXP: spmatrix | ndarray | DataFrame, metadata_regions: DataFrame, metadata_genes: DataFrame, metadata_cell: DataFrame, menr: Mapping[str, Mapping[str, Any]], dr_cell: Mapping[str, iterable] = None, dr_region: Mapping[str, iterable] = None, uns: Mapping[str, Any] = {})[source]#
An object containing: gene expression, chromatin accessbility and motif enrichment data.
SCENICPLUSstores the data matricesX_ACC(chromatin accessbility) andX_EXP(gene expression) together with region annotationmetadata_regions, gene annotationmetadata_genes, cell annotationmetadata_celland motif enrichment datamenr.Use create_SCENICPLUS_object to generate instances of this class.
- Parameters:
- X_ACC: sparse.spmatrix, np.ndarray, pd.DataFrame
A #regions x #cells data matrix
- X_EXP: sparse.spmatrix, np.ndarray, pd.DataFrame
A #cells x #genes data matrix
- metadata_regions: pd.DataFrame
A
pandas.DataFramecontaining region metadata annotation of length #regions- metadata_genes: pd.DataFrame
A
pandas.DataFramecontaining gene metadata annotation of length #genes- metadata_cell: pd.DataFrame
A
pandas.DataFramecontaining cell metadata annotation of lenght #cells- menr: dict
A Dict containing motif enrichment results for topics of differentially accessbile regions (DARs), generate by pycistarget. Should take the form {‘region_set_name1’: {region_set1: result, ‘region_set2’: result}, ‘region_set_name2’: {‘region_set1’: result, ‘region_set2’: result}, ‘topic’: {‘topic1’: result, ‘topic2’: result}} region set names, which aren’t topics, should be columns in the
metadata_celldataframe- dr_cell: dict
A Dict containing dimmensional reduction coordinates of cells.
- dr_region: dict
A Dict containing dimmensional reduction coordinates of regions.
- Attributes:
- n_cells: int
Returns number of cells.
- n_genes: int
Returns number of genes.
- n_regions: int
Returns number of regions.
- cell_names: List[str]
Returns cell names
- gene_names: List[str]
Returns gene names
- region_names: List[str]
Returns region names
Methods
add_cell_data(cell_data)Add cell metadata
add_gene_data(gene_data)Add gene metadata
add_region_data(region_data)Add region metadata
subset([cells, regions, genes, return_copy])Subset object
to_df(layer)Generate a
DataFrame.- add_cell_data(cell_data: DataFrame)[source]#
Add cell metadata
- Parameters:
- cell_data: pd.DataFrame
A
DataFramecontaining cell metdata indexed with cell barcodes.
- add_gene_data(gene_data: DataFrame)[source]#
Add gene metadata
- Parameters:
- gene_data: pd.DataFrame
A
DataFramecontaining gene metadata indexed with gene names.
- add_region_data(region_data: DataFrame)[source]#
Add region metadata
- Parameters:
- region_data: pd.DataFrame
A
DataFramecontaining region metadata indexed with region names.
- subset(cells=None, regions=None, genes=None, return_copy=False)[source]#
Subset object
- Parameters:
- cells: List[str]
A list of cells to keep default: None
- regions: List[str]
A list of regions to keep default: None
- genes: List[str]
A list of genes to keep default:None
- return_copy: bool
A boolean specifying wether to update the object (False) or return a copy (True)
- scenicplus.scenicplus_class.create_SCENICPLUS_object(GEX_anndata: AnnData, cisTopic_obj: CistopicObject, menr: Mapping[str, Mapping[str, Any]], multi_ome_mode: bool = True, nr_metacells: int | Mapping[str, int] = None, nr_cells_per_metacells: int | Mapping[str, int] = 10, meta_cell_split: str = '_', key_to_group_by: str = None, imputed_acc_obj: CistopicImputedFeatures = None, imputed_acc_kwargs: Mapping[str, Any] = {'scale_factor': 1000000}, normalize_imputed_acc: bool = False, normalize_imputed_acc_kwargs: Mapping[str, Any] = {'scale_factor': 10000}, cell_metadata: DataFrame = None, region_metadata: DataFrame = None, gene_metadata: DataFrame = None, bc_transform_func: Callable = None, ACC_prefix: str = 'ACC_', GEX_prefix: str = 'GEX_') SCENICPLUS[source]#
Function to create instances of
SCENICPLUS- Parameters:
- GEX_anndata: sc.AnnData
An instance of
AnnDatacontaining gene expression data and metadata.- cisTopic_obj: CistopicObject
An instance of
pycisTopic.cistopic_class.CistopicObjectcontaining chromatin accessibility data and metadata.- menr: dict
A dict mapping annotations to motif enrichment results
- multi_ome_mode: bool
A boolean specifying wether data is multi-ome (i.e. combined scATAC-seq and scRNA-seq from the same cell) or not default: True
- nr_metacells: int
For non multi_ome_mode, use this number of meta cells to link scRNA-seq and scATAC-seq If this is a single integer the same number of metacells will be used for all annotations. This can also be a mapping between an annotation and the number of metacells per annotation. default: None
- nr_cells_per_metacells: int
For non multi_ome_mode, use this number of cells per metacell to link scRNA-seq and scATAC-seq. If this is a single integer the same number of cells will be used for all annotations. This can also be a mapping between an annotation and the number of cells per metacell per annotation. default: 10
- meta_cell_split: str
Character which is used as seperator in metacell names default: ‘_’
- key_to_group_by: str
For non multi_ome_mode, use this cell metadata key to generate metacells from scRNA-seq and scATAC-seq. Key should be common in scRNA-seq and scATAC-seq side default: None
- imputed_acc_obj: CistopicImputedFeatures
An instance of
CistopicImputedFeaturescontaining imputed chromatin accessibility. default: None- imputed_acc_kwargs: dict
Dict with keyword arguments for imputed chromatin accessibility. default: {‘scale_factor’: 10**6}
- normalize_imputed_acc: bool
A boolean specifying wether or not to normalize imputed chromatin accessbility. default: False
- normalize_imputed_acc_kwargs: dict
Dict with keyword arguments for normalizing imputed accessibility. default: {‘scale_factor’: 10 ** 4}
- cell_metadata: pd.DataFrame
An instance of
DataFramecontaining extra cell metadata default: None- region_metadata: pd.DataFrame
An instance of
DataFramecontaining extra region metadata default: None- gene_metadata: pd.DataFrame
An instance of
DataFramecontaining extra gene metadata default: None- bc_transform_func: func
A function used to transform gene expression barcode layout to chromatin accessbility layout. default: None
- ACC_prefix: str
String prefix to add to cell metadata coming from cisTopic_obj
- GEX_prefix: str
String prefix to add to cell metadata coming from GEX_anndata
Examples
>>> scplus_obj = create_SCENICPLUS_object(GEX_anndata = rna_anndata, cisTopic_obj = cistopic_obj, menr = motif_enrichment_dict)
- scenicplus.scenicplus_class.mudata_to_scenicplus(mdata: MuData, path_to_cistarget_h5: str | None = None, path_to_dem_h5: str | None = None) SCENICPLUS[source]#
Convert a MuData object to a SCENICPLUS object.
- Parameters:
- mdata: MuData
An instance of
MuDatacreated after running the scenicplus workflow.- path_to_cistarget_h5: str
Path to the h5 file containing cistarget results. default: None
- path_to_dem_h5: str
Path to the h5 file containing dem results. default: None
- Returns:
- SCENICPLUS
An instance of
SCENICPLUS
Preprocessing#
Filter outlier genes and regions.
SCENIC+ semi-automated workflow using wrapper functions#
pycistarget wrapper#
Wrapper functions to run motif enrichment analysis using pycistarget
After sets of regions have been defined (e.g. topics or DARs). The complete pycistarget workflo can be run using a single function.
this function will run cistarget based and DEM based motif enrichment analysis with or without promoter regions.
- scenicplus.wrappers.run_pycistarget.run_pycistarget(region_sets: Dict[str, PyRanges], species: str, save_path: str, custom_annot: DataFrame = None, save_partial: bool = False, ctx_db_path: str = None, dem_db_path: str = None, run_without_promoters: bool = False, biomart_host: str = 'http://www.ensembl.org', promoter_space: int = 500, ctx_auc_threshold: float = 0.005, ctx_nes_threshold: float = 3.0, ctx_rank_threshold: float = 0.05, dem_log2fc_thr: float = 0.5, dem_motif_hit_thr: float = 3.0, dem_max_bg_regions: int = 500, annotation: List[str] = ['Direct_annot', 'Orthology_annot'], motif_similarity_fdr: float = 1e-06, path_to_motif_annotations: str = None, annotation_version: str = 'v9', n_cpu: int = 1, _temp_dir: str = None, exclude_motifs: str = None, exclude_collection: List[str] = None, **kwargs)[source]#
Wrapper function for pycistarget
- Parameters:
region_sets (Mapping[str, pr.PyRanges]) – A dictionary of PyRanges containing region coordinates for the region sets to be analyzed.
species (str) – Species from which genomic coordinates come from, options are: homo_sapiens, mus_musculus, drosophila_melanogaster and gallus_gallus.
save_path (str) – Directory in which to save outputs.
custom_annot (pd.DataFrame) –
pandas DataFrame with genome annotation for custom species (i.e. for a species other than homo_sapiens, mus_musculus, drosophila_melanogaster or gallus_gallus). This DataFrame should (minimally) look like the example below, and only contains protein coding genes: >>> custom_annot
Chromosome Start Strand Gene Transcript_type
8053 chrY 22490397 1 PRY protein_coding 8153 chrY 12662368 1 USP9Y protein_coding 8155 chrY 12701231 1 USP9Y protein_coding 8158 chrY 12847045 1 USP9Y protein_coding 8328 chrY 22096007 -1 PRY2 protein_coding … … … … … … 246958 chr1 181483738 1 CACNA1E protein_coding 246960 chr1 181732466 1 CACNA1E protein_coding 246962 chr1 181776101 1 CACNA1E protein_coding 246963 chr1 181793668 1 CACNA1E protein_coding 246965 chr1 203305519 1 BTG2 protein_coding
[78812 rows x 5 columns]
save_partial (bool=False) – Whether to save the individual analyses as pkl. Useful to run analyses in chunks or add new settings.
ctx_db_path (str = None) – Path to cistarget database containing rankings of motif scores
dem_db_path (str = None) – Path to dem database containing motif scores
run_without_promoters (bool = False) – Boolean specifying wether the analysis should also be run without including promoter regions.
biomart_host (str = ‘http://www.ensembl.org’) – url to biomart host, make sure this host matches your assembly
promoter_space (int = 500) – integer defining space around the TSS to consider as promoter
ctx_auc_threshold (float = 0.005) – The fraction of the ranked genome to take into account for the calculation of the Area Under the recovery Curve
ctx_nes_threshold (float = 3.0) – The Normalized Enrichment Score (NES) threshold to select enriched features.
ctx_rank_threshold (float = 0.05) – The total number of ranked genes to take into account when creating a recovery curve.
dem_log2fc_thr (float = 0.5) – Log2 Fold-change threshold to consider a motif enriched.
dem_motif_hit_thr (float = 3.0) – Minimul mean signal in the foreground to consider a motif enriched.
dem_max_bg_regions (int = 500) – Maximum number of regions to use as background. When set to None, all regions are used
annotation (List[str] = ['Direct_annot', 'Orthology_annot']) – Annotation to use for forming cistromes. It can be ‘Direct_annot’ (direct evidence that the motif is linked to that TF), ‘Motif_similarity_annot’ (based on tomtom motif similarity), ‘Orthology_annot’ (based on orthology with a TF that is directly linked to that motif) or ‘Motif_similarity_and_Orthology_annot’.
path_to_motif_annotations (str = None) – Path to motif annotations. If not provided, they will be downloaded from https://resources.aertslab.org based on the specie name provided (only possible for mus_musculus, homo_sapiens and drosophila_melanogaster).
annotation_version (str = 'v9') – Motif collection version.
n_cpu (int = 1) – Number of cores to use.
_temp_dir (str = None) – temp_dir to use for ray.
exclude_motifs (str = None) – Path to csv file containing motif to exclude from the analysis.
exclude_collection (List[str] = None) – List of strings identifying which motif collections to exclude from analysis.
SCENIC+ wrapper#
Tools for non-automated workflow#
Cistromes#
Enhancer-to-gene linking#
Link enhancers to genes based on co-occurence of chromatin accessbility of the enhancer and gene expression.
Both linear methods (spearman or pearson correlation) and non-linear methods (random forrest or gradient boosting) are used to link enhancers to genes.
The correlation methods are used to seperate regions which are infered to have a positive influence on gene expression (i.e. positive correlation) and regions which are infered to have a negative influence on gene expression (i.e. negative correlation).
- scenicplus.enhancer_to_gene.calculate_regions_to_genes_relationships(df_exp_mtx: DataFrame, df_acc_mtx: DataFrame, search_space: DataFrame, temp_dir: Path, mask_expr_dropout: bool = False, importance_scoring_method: Literal['RF', 'ET', 'GBM'] = 'GBM', importance_scoring_kwargs: dict = {'learning_rate': 0.01, 'max_features': 0.1, 'n_estimators': 500}, correlation_scoring_method: Literal['PR', 'SR'] = 'SR', n_cpu: int = 1, add_distance: bool = True)[source]#
# TODO: add docstrings
- scenicplus.enhancer_to_gene.export_to_UCSC_interact(scplus_obj: SCENICPLUS, species: str, outfile: str, region_to_gene_key: str = ' region_to_gene', pbm_host: str = 'http://www.ensembl.org', bigbed_outfile: str = None, path_bedToBigBed: str = None, assembly: str = None, ucsc_track_name: str = 'region_to_gene', ucsc_description: str = 'interaction file for region to gene', cmap_neg: str = 'Reds', cmap_pos: str = 'Greens', key_for_color: str = 'importance', vmin: int = 0, vmax: int = 1, scale_by_gene: bool = True, subset_for_eRegulons_regions: bool = True, eRegulons_key: str = 'eRegulons') DataFrame[source]#
Exports interaction dataframe to UCSC interaction file and (optionally) UCSC bigInteract file.
- Parameters:
- scplus_obj: SCENICPLUS
An instance of class scenicplus_class.SCENICPLUS containing region to gene links in .uns.
- species: str
Species corresponding to your datassets (e.g. hsapiens)
- outfile: str
Path to output file
- region_to_gene_key: str =’ region_to_gene’
Key in scplus_obj.uns.keys() under which to find region to gene links.
- pbm_host:str = ‘http://www.ensembl.org’
Url of biomart host relevant for your assembly.
- bigbed_outfile:str = None
Path to which to write the bigbed output.
- path_bedToBigBed: str= None
Path to bedToBigBed program, used to convert bed file to bigbed format. Can be downloaded from http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/bedToBigBed
- assembly: str = None
String identifying the assembly of your dataset (e.g. hg39).
- ucsc_track_name: str = ‘region_to_gene’
Name of the exported UCSC track
- ucsc_description: str = ‘interaction file for region to gene’
Description of the exported UCSC track
- cmap_neg: str = ‘Reds’
Matplotlib colormap used to color negative region to gene links.
- cmap_pos: str = ‘Greens’
Matplotlib colormap used to color positive region to gene links.
- key_for_color: str = ‘importance’
Key pointing to column in region to gene links used to map cmap colors to.
- vmin: int = 0
vmin of region to gene link colors.
- vmax: int = 1
vmax of region to gene link colors.
- scale_by_gene: bool = True
Boolean specifying wether to scale importance scores of regions linking to the same gene from 0 to 1
- subset_for_eRegulons_regions: bool = True
Boolean specifying wether or not to subset region to gene links for regions and genes in eRegulons.
- eRegulons_key: str = ‘eRegulons’
key in scplus_obj.uns.keys() under which to find eRegulons.
- Returns:
- pd.DataFrame with region to gene links formatted in the UCSC interaction format.
TF-to-gene linking#
eGRN building#
eRegulon Class#
eRegulon class stores the transcription factor together with its target regions and genes.
- class scenicplus.grn_builder.modules.eRegulon(transcription_factor: str, cistrome_name: str, is_extended: bool, regions2genes: List[namedtuple], context=frozenset({}), in_leading_edge: List[bool] = None, gsea_enrichment_score: float = None, gsea_pval: float = None, gsea_adj_pval: float = None)[source]#
An eRegulon is a gene signature that defines the target regions and genes of a Transcription Factor (TF).
- Parameters:
- transcription_factor
A string specifying the transcription factor of the eRegulon
- cistrome_name
A string specifying the cistrome name
- is_extended
A boolean specifying wether the cistromes comes from an extended annotation or not
- regions2genes
A list of named tuples containing information on region-to-gene links (region, gene, importance score, correlation coefficient)
- context
The context in which this eRegulon was created (this can be different threshold, activating / repressing, …). default: frozenset()
- in_leading_edge
A list specifying which genes are in the leading edge of a gsea analysis. default: None
- gsea_enrichment_score
A float containing the enrichment score of a gsea analysis. default: None
- gsea_pval
A float containing the p value of a gsea analysis. default: None
- gsea_adj_pval
A float containing an adjusted p value of a gsea analysis. default: None
- Attributes:
target_genesReturn target genes of this eRegulon.
target_regionsReturn target regions of this eRegulon.
n_target_genesReturn number of target genes.
n_target_regionsReturn number of target regions.
Methods
subset_leading_edge([inplace])Subset eReglon on leading edge.
See also
scenicplus.grn_builder.modules.create_emodules
- property n_target_genes#
Return number of target genes.
- property n_target_regions#
Return number of target regions.
- subset_leading_edge(inplace=True)[source]#
Subset eReglon on leading edge.
- Parameters:
inplace – if set to True update eRegulon else return new eRegulon after subset.
- property target_genes#
Return target genes of this eRegulon.
- property target_regions#
Return target regions of this eRegulon.
- scenicplus.grn_builder.modules.merge_emodules(SCENICPLUS_obj: SCENICPLUS = None, e_modules: list = None, e_modules_key: str = 'eRegulons', rho_dichotomize: bool = True, key_to_add: str = 'eRegulons', inplace: bool = True)[source]#
Function to merge list of
eRegulon- Parameters:
- SCENICPLUS_obj
An instance of :class: ~scenicplus.scenicplus_class.SCENICPLUS, containing eRegulons in slot .uns. default: None
- e_modules
A list of
eRegulon. default: None- e_modules_key
A string key specifying where to find a list of
eRegulonunder .uns slot of`SCENICPLUS_obj`. default: “eRegulons”- rho_dichotomize
A boolean specifying wether or not to split region-to-gene links based on postive/negative correlation coefficients. default: True
- key_to_add
A string key specifying where to store the list of merged
eRegulonin the .uns slot of SCENICPLUS_obj.- inplace
A boolean if set to True update the SCENICPLUS_obj otherwise return list of merged
eRegulon
GSEA based approach#
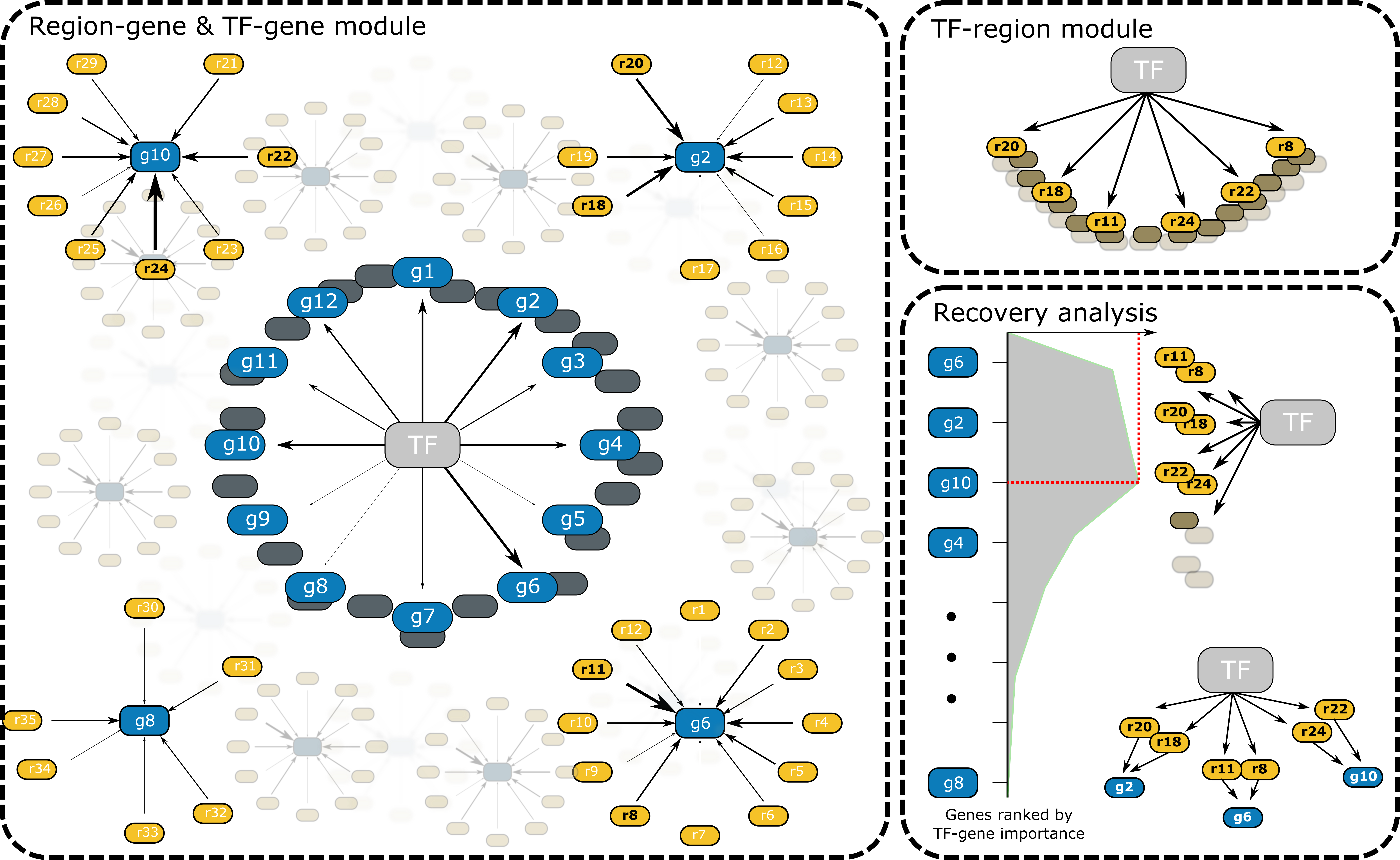
Generate enhancer drive GRNs (eGRS) using the GSEA approach.
Using this approach we will test if the gene set obtained from region-to-gene links, where the regions have a high score for a motif of a certain TF (region indicated in black in the diagram: r8, r11, r18, r20, r22, r24), are enriched in the top of the ranking based on the TF-to-gene links of the same TF (bottom right panel).
Only genes, from the set, and the regions linked to these genes in the top of the ranking (i.e. leading edge) will be kept.
This aproach is done seperatly for positive and negative TF and region to gene links.
Generating following four combinations:
TF-to-gene relationship |
region-to-gene relationship |
biological role |
|---|---|---|
positive (+) |
positive (+) |
TF opens chromatin and activates gene expression |
positive (+) |
negative (-) |
When the TF is expressed the target gene is also expressed but the regions linked to the gene are closed. |
negative (-) |
positive (+) |
When the TF is expressed the target gene is not expressed. When the target gene is expressed, regions linked to this gene are open. TF could be a chromatin closing repressor. |
negative (-) |
negative (-) |
When the TF is expressed the target gene is not expressed. When the target gene is expressed, regions linked to this gene are closed. |
Left panel indicates the TF-to-gene links (blue) and region-to-gene links (yellow). Witdh of arrows correspond the strength of the connections based on non-linear regression.
Top right panel indicates regions with a high score for a motif of a TF
Bottom right panel shows GSEA analysis, with on the left genes ranked by TF-to-gene connection strength and on the right the gene-set obtained from region-to-gene links. In the diagram: g2, g6, and g10 are located in the top of the TF-to-gene ranking (i.e. leading edge), only these genes and the regions linked to these genes: r20, r18, r11, r8, r24 and r22 will be kept.
Downstream analysis, export and plotting#
Marker genes and regions#
eRegulon enrichment in cells#
Score eRegulon target genes and regions in cells using AUC algorithm.
- scenicplus.eregulon_enrichment.binarize_AUC(scplus_obj: SCENICPLUS, auc_key: str | None = 'eRegulon_AUC', out_key: str | None = 'eRegulon_AUC_thresholds', signature_keys: List[str] | None = ['Gene_based', 'Region_based'], n_cpu: int | None = 1)[source]#
Binarize eRegulons using AUCell
- Parameters:
- scplus_obj: `class::SCENICPLUS`
A SCENICPLUS object with eRegulons AUC.
- auc_key: str, optional
Key where the AUC values are stored
- out_key: str, optional
Key where the AUCell thresholds will be stored (in scplus_obj.uns)
- signature_keys: List, optional
Keys to extract AUC values from. Default: [‘Gene_based’, ‘Region_based’]
- n_cpu: int
The number of cores to use. Default: 1
- scenicplus.eregulon_enrichment.get_eRegulons_as_signatures(eRegulons: DataFrame) Dict[str, Dict[str, List[str]]][source]#
- scenicplus.eregulon_enrichment.rank_data(df: DataFrame, axis: Literal[0, 1] = 1, seed: int = 123) ranked_data[source]#
dimensionality reduction#
eRegulon specificity score (eRSS)#
Calculate the specificty of eRegulons in clusters of cells.
Calculates the distance between the real distribution of eRegulon AUC values and a fictional distribution where the eRegulon is only expressed/accessible in cells of a certain cluster.
- scenicplus.RSS.plot_rss(data_matrix: DataFrame, top_n: int = 5, selected_groups: List[str] = None, num_columns: int = 1, figsize: Tuple[float, float] = (6.4, 4.8), fontsize: int = 12, save: str = None)[source]#
Plot RSS values per group
- Parameters:
data_matrix (class::pd.DataFrame) – A pandas dataframe with RSS scores per variable.
top_n (int, optional) – Number of top eRegulons to highlight.
selected_groups (List, optional) – Groups to plot. Default: None (all)
num_columns (int, optional) – Number of columns for multiplotting
figsize (tuple, optional) – Size of the figure. If num_columns is 1, this is the size for each figure; if num_columns is above 1, this is the overall size of the figure (if keeping default, it will be the size of each subplot in the figure). Default: (6.4, 4.8)
fontsize (int, optional) – Size of the eRegulons names in plot.
save (str, optional) – Path to save plot. Default: None.
- scenicplus.RSS.regulon_specificity_scores(scplus_mudata: MuData | ScenicPlusMuData, variable: str, modalities: list, selected_regulons: List[int] = None)[source]#
Calculate the Regulon Specificty Scores (RSS). [doi: 10.1016/j.celrep.2018.10.045]
- Parameters:
scplus_mudata (class::MuData or ‘class::ScenicPlusMuData’) – A MuData object with eRegulons AUC computed.
variable (str) – Variable to calculate the RSS values for.
modalities (List,) – A list of modalities to calculate RSS values for.
selected_regulons (List, optional) – Regulons to calculate RSS values for.
- scenicplus.RSS.regulon_specificity_scores_df(data_matrix: DataFrame, variable_matrix: Series)[source]#
Calculate the Regulon Specificty Scores (RSS). [doi: 10.1016/j.celrep.2018.10.045]
- Parameters:
data_matrix ('class::pd.DataFrame`) – A pandas dataframe containing regulon scores per cell.
variable_matrix ('class::pd.Series') – A pandas series with an annotation per cell.
Triplet score#
Calculate the TF-region-gene triplet ranking
The triplet ranking is the aggregated ranking of TF-to-region scores, region-to-gene scores and TF-to-region scores. The TF-to-gene and TF-to-region scores are defined as the feature importance scores for predicting gene expression from resp. TF expression and region accessibility. The TF-to-region score is defined as the maximum motif-score-rank for a certain region across all motifs annotated to the TF of interest.
Network#
export eRegulons to eGRN network and plot.
- scenicplus.networks.concentrical_layout(G, dist_genes=1, dist_TF=0.1)[source]#
Generate custom concentrical layout
- Parameters:
G (Graph) – A networkx graph
dist_genes (int, optional) – Distance from the regions to the genes
dist_TF – Distance from the TF to the regions
- scenicplus.networks.create_nx_graph(nx_tables: Dict, use_edge_tables: List = ['TF2R', 'R2G'], color_edge_by: Dict = {}, transparency_edge_by: Dict = {}, width_edge_by: Dict = {}, color_node_by: Dict = {}, transparency_node_by: Dict = {}, size_node_by: Dict = {}, shape_node_by: Dict = {}, label_size_by: Dict = {}, label_color_by: Dict = {}, layout: str = 'concentrical_layout', lc_dist_genes: float = 0.8, lc_dist_TF: float = 0.1, scale_position_by: float = 250)[source]#
Format node/edge feature tables into a graph
- Parameters:
nx_tables (Dict) – Dictionary with node/edge feature tables as produced by create_nx_tables
use_edge_tables (List, optional) – List of edge tables to use
color_edge_by (Dict, optional) – A dictionary containing for a given edge key the variable and color map to color edges by. If the variable is categorical, the entry ‘categorical_color’ can be provided as a dictionary with category: color. If it is a continuous variable a color map can be provided as continuous_color and entried v_max and v_min can be provided to control the min and max values of the scale. Alternatively, one fixed color can use by using ‘fixed_color’ as variable, alterntively adding an entry fixed_color: color to the dictionary.
transparency_edge_by (Dict, optional) – A dictionary containing for a given edge key the variable and the max and min alpha values. The variable name has to be provided (only continuous variables accepted), together with v_max/v_mix parameters if desired. Alternatively, one fixed alpha can use by using ‘fixed_alpha’ as variable, alterntively adding an entry fixed_alpha: size to the dictionary.
width_edge_by (Dict, optional) – A dictionary containing for a given edge key the variable and the max and min sizes. The variable name has to be provided (only continuous variables accepted), together with max_size/min_size parameters if desired. Alternatively, one fixed size can use by using ‘fixed_size’ as variable, alterntively adding an entry fixed_size: size to the dictionary.
color_node_by (Dict, optional) – A dictionary containing for a given node key the variable and color map to color edges by. If the variable is categorical, the entry ‘categorical_color’ can be provided as a dictionary with category: color. If it is a continuous variable a color map can be provided as continuous_color and entried v_max and v_min can be provided to control the min and max values of the scale. Alternatively, one fixed color can use by using ‘fixed_color’ as variable, alterntively adding an entry fixed_color: color to the dictionary.
transparency_node_by (Dict, optional) – A dictionary containing for a given node key the variable and the max and min alpha values. The variable name has to be provided (only continuous variables accepted), together with v_max/v_mix parameters if desired. Alternatively, one fixed alpha can use by using ‘fixed_alpha’ as variable, alterntively adding an entry fixed_alpha: size to the dictionary.
size_node_by (Dict, optional) – A dictionary containing for a given node key the variable and the max and min sizes. The variable name has to be provided (only continuous variables accepted), together with max_size/min_size parameters if desired. Alternatively, one fixed size can use by using ‘fixed_size’ as variable, alterntively adding an entry fixed_size: size to the dictionary.
shape_node_by (Dict, optional) – A dictionary containing for a given node key the variable and shapes. The variable name has to be provided (only categorical variables accepted). Alternatively, one fixed shape can use by using ‘fixed_shape’ as variable, alterntively adding an entry fixed_shape: size to the dictionary.
label_size_by (Dict, optional) – A dictionary containing for a given node key the variable and the max and min sizes. The variable name has to be provided (only continuous variables accepted), together with max_size/min_size parameters if desired. Alternatively, one fixed size can use by using ‘fixed_label_size’ as variable, alterntively adding an entry fixed_label_size: size to the dictionary.
label_color_by (Dict, optional) – A dictionary containing for a given node key the variable and a color dictionary. The variable name has to be provided (only categorical variables accepted), together with a color dictionary if desired. Alternatively, one fixed color can use by using ‘fixed_label_color’ as variable, alterntively adding an entry fixed_label_color: size to the dictionary.
layout (str, optional) – Layout to use. Options are: ‘concentrical_layout’ (SCENIC+ custom layout) or kamada_kawai_layout (from networkx).
lc_dist_genes (float, optional) – Distance between regions and genes. Only used if using concentrical_layout.
lc_dist_TF (float, optional) – Distance between TF and regions. Only used if using concentrical_layout.
scale_position_by (int, optional) – Value to scale positions for visualization in pyvis.
- scenicplus.networks.create_nx_tables(scplus_obj: SCENICPLUS, eRegulon_metadata_key: str = 'eRegulon_metadata', subset_eRegulons: List = None, subset_regions: List = None, subset_genes: List = None, add_differential_gene_expression: bool = False, add_differential_region_accessibility: bool = False, differential_variable: List = [])[source]#
A function to format eRegulon data into tables for plotting eGRNs.
- Parameters:
scplus_obj (SCENICPLUS) – A SCENICPLUS object with eRegulons
eRegulon_metadata_key (str, optional) – Key where the eRegulon metadata dataframe is stored
subset_eRegulons (list, optional) – List of eRegulons to subset
subset_regions (list, optional) – List of regions to subset
subset_genes (list, optional) – List of genes to subset
add_differential_gene_expression (bool, optional) – Whether to calculate differential gene expression logFC for a given variable
add_differential_region_accessibility (bool, optional) – Whether to calculate differential region accessibility logFC for a given variable
differential_variable (list, optional) – Variable to calculate differential gene expression or region accessibility.
- scenicplus.networks.export_to_cytoscape(G, pos, out_file: str, pos_scaling_factor: int = 200, size_scaling_factor: int = 1)[source]#
A function to export to cytoscape :param G: A networkx graph. :type G: Graph :param Pos: generated by running create_nx_graph. :type Pos: coordinates of graph nodes :param out_file: Path to wich to save the export. :type out_file: str :param pos_scaling_factor: Factor by which to scale the graph node coordinates. :type pos_scaling_factor: int, optional :param size_scaling_factor: Factor by which tos cale the graph node sizes. :type size_scaling_factor: int, optional
Correlation plot#
Plot correlation and overlap of eRegulons
- scenicplus.plotting.correlation_plot.correlation_heatmap(scplus_obj: SCENICPLUS, auc_key: str | None = 'eRegulon_AUC', signature_keys: List[str] | None = ['Gene_based', 'Region_based'], scale: bool | None = False, linkage_method: str | None = 'average', fcluster_threshold: float | None = 0.1, selected_regulons: List[int] | None = None, cmap: str | None = 'viridis', plotly_height: int | None = 1000, fontsize: int | None = 3, save: str | None = None, use_plotly: int | None = True, figsize: Tuple[int, int] | None = (20, 20))[source]#
Plot correlation between eRegulons enrichment,
- Parameters:
scplus_obj (class::SCENICPLUS) – A SCENICPLUS object with eRegulons AUC computed.
auc_key (str, optional) – Key to extract AUC values from. Default: ‘eRegulon_AUC’
signature_keys (List, optional) – Keys to extract AUC values from. Default: [‘Gene_based’, ‘Region_based’]
scale (bool, optional) – Whether to scale the enrichments prior to the dimensionality reduction. Default: False
linkage_method (str, optional) – Linkage method to use for clustering. See scipy.cluster.hierarchy.linkage.
fcluster_threshold (float, optional) – Threshold to use to divide hierarchical clustering into clusters. See scipy.cluster.hierarchy.fcluster.
selected_regulons (list, optional) – A list with selected regulons to be used for clustering. Default: None (use all regulons)
cmap (str or 'matplotlib.cm', optional) – For continuous variables, color map to use for the legend color bar. Default: cm.viridis
plotly_height (int, optional) – Height of the plotly plot. Width will be adjusted accordingly
fontsize (int, optional) – Labels fontsize
save (str, optional) – Path to save heatmap as file
use_plotly (bool, optional) – Use plotly or seaborn to generate the image
figsize (tupe, optional) – Matplotlib figsize, used only if use_plotly == False
- scenicplus.plotting.correlation_plot.fisher_exact_test_heatmap(scplus_obj, gene_or_region_based: str = 'Gene_based', signature_key: str | None = 'eRegulon_signatures', selected_regulons: List[int] | None = None, linkage_method: str | None = 'average', fcluster_threshold: float | None = 0.1, cmap: str | None = 'viridis', plotly_height: int | None = 1000, fontsize: int | None = 3, save: str | None = None, use_plotly: int | None = True, figsize: Tuple[int, int] | None = (20, 20), vmin=None, vmax=None, return_data=False)[source]#
Plot jaccard index of regions/genes
- Parameters:
scplus_obj (class::SCENICPLUS) – A SCENICPLUS object with eRegulon signatures.
method (str) – Wether to use Jaccard (jaccard) or normalized intersection (intersect) as metric
gene_or_region_based (str) – Gene_based or Region_based eRegulon signatures to use.
signature_key (List, optional) – Key to extract eRegulon signatures from
selected_regulons (list, optional) – A list with selected regulons to be used for clustering. Default: None (use all regulons)
linkage_method (str, optional) – Linkage method to use for clustering. See scipy.cluster.hierarchy.linkage.
fcluster_threshold (float, optional) – Threshold to use to divide hierarchical clustering into clusters. See scipy.cluster.hierarchy.fcluster.
cmap (str or 'matplotlib.cm', optional) – For continuous variables, color map to use for the legend color bar. Default: cm.viridis
plotly_height (int, optional) – Height of the plotly plot. Width will be adjusted accordingly
fontsize (int, optional) – Labels fontsize
save (str, optional) – Path to save heatmap as file
use_plotly (bool, optional) – Use plotly or seaborn to generate the image
figsize (tupe, optional) – Matplotlib figsize, used only if use_plotly == False
return_data (boolean, optional) – Return data
plot_dendrogram (boolean, optional)
- scenicplus.plotting.correlation_plot.jaccard_heatmap(scplus_obj: SCENICPLUS, method: str = 'jaccard', gene_or_region_based: str = 'Gene_based', signature_key: str | None = 'eRegulon_signatures', selected_regulons: List[int] | None = None, linkage_method: str | None = 'average', fcluster_threshold: float | None = 0.1, cmap: str | None = 'viridis', plotly_height: int | None = 1000, fontsize: int | None = 3, save: str | None = None, use_plotly: int | None = True, figsize: Tuple[int, int] | None = (20, 20), vmin=None, vmax=None, return_data=False)[source]#
Plot jaccard index of regions/genes
- Parameters:
scplus_obj (class::SCENICPLUS) – A SCENICPLUS object with eRegulon signatures.
method (str) – Wether to use Jaccard (jaccard) or normalized intersection (intersect) as metric
gene_or_region_based (str) – Gene_based or Region_based eRegulon signatures to use.
signature_key (List, optional) – Key to extract eRegulon signatures from
selected_regulons (list, optional) – A list with selected regulons to be used for clustering. Default: None (use all regulons)
linkage_method (str, optional) – Linkage method to use for clustering. See scipy.cluster.hierarchy.linkage.
fcluster_threshold (float, optional) – Threshold to use to divide hierarchical clustering into clusters. See scipy.cluster.hierarchy.fcluster.
cmap (str or 'matplotlib.cm', optional) – For continuous variables, color map to use for the legend color bar. Default: cm.viridis
plotly_height (int, optional) – Height of the plotly plot. Width will be adjusted accordingly
fontsize (int, optional) – Labels fontsize
save (str, optional) – Path to save heatmap as file
use_plotly (bool, optional) – Use plotly or seaborn to generate the image
figsize (tupe, optional) – Matplotlib figsize, used only if use_plotly == False
return_data (boolean, optional) – Return data
plot_dendrogram (boolean, optional)
Coverage plot#
Plot chromatin accessibility profiles and region to gene arcs.
- scenicplus.plotting.coverageplot.coverage_plot(SCENICPLUS_obj: SCENICPLUS, bw_dict: Mapping[str, str], region: str, genes_violin_plot: str | List = None, genes_arcs: str | List = None, gene_height: int = 1, exon_height: int = 4, meta_data_key: str = None, pr_consensus_bed: PyRanges = None, region_bed_height: int = 1, pr_gtf: PyRanges = None, pr_interact: PyRanges = None, bw_ymax: float = None, color_dict: Mapping[str, str] = None, cmap='tab20', plot_order: list = None, figsize: tuple = (6, 8), fontsize_dict={'bigwig_label': 9, 'bigwig_tick_label': 5, 'gene_label': 9, 'title': 12, 'violinplots_xlabel': 9, 'violinplots_ylabel': 9}, gene_label_offset=3, arc_rad=0.5, arc_lw=1, cmap_violinplots='Greys', violinplots_means_color='black', violinplots_edge_color='black', violoinplots_alpha=1, width_ratios_dict={'bigwig': 3, 'violinplots': 1}, height_ratios_dict={'arcs': 5, 'bigwig_violin': 1, 'custom_ax': 2, 'genes': 0.5}, sort_vln_plots: bool = False, add_custom_ax: int = None) Figure[source]#
Inspired by: https://satijalab.org/signac/reference/coverageplot
Generates a figure showing chromatin accessibility coverage tracks, gene expression violin plots and region to gene interactions.
- Parameters:
- SCENICPLUS_obj
An instance of class ~scenicplus.scenicplus_class.SCENICPLUS.
- bw_dict
A dict containing celltype annotations/cell groups as keys and paths to bigwig files as values.
- region
A string specifying the region of interest, in chr:start-end format.
- genes_violin_plot
A list or string specifying for which gene(s) to plot gene expression violin plots. default: None
- genes_arcs
A list or string specifying for which gene(s) to plot arcs default: None (i.e. all genes in window)
- gene_height
An int specifying the size of non-exon parts of a gene (shown underneath the coverage plot). default: 1
- exon_height
An int specifying the size of nexon parts of a gene (shown underneath the coverage plot). default: 2
- meta_data_key
A key specifying were to find annotations corresponding to the keys in bw_dict in SCENICPLUS_obj.metadata_cell default: None
- pr_consensus_bed
An instance of class pyranges.PyRanges containing consensus peaks. Names of these peaks should correspond to annotations. default: None
- region_bed_height
An int specifying with which height to draw lines corresponding to regions in pr_consensus_bed default: 1
- pr_gtf
An instance of class pyranges.PyRanges containing gtf-formatted information about gene structure. default: None
- pr_interact
An instance of class pyranges.PyRanges containing region to gene link information in ucsc interact format (use scenicplus.utils.get_interaction_pr to create such an object). default: None
- bw_ymax
A float specifying the maximum height at which to draw coverage plots. default: None
- color_dict
A dict specifying colors for each key in bw_dict. default: None
- cmap
A string specifying a matplotlib.cm colormap to use for coloring coverage plots. default: ‘tab20’
- plot_order
A list specifying the order to use for plotting coverage plots and violin plots. default: None
- figsize
A tuple specifying the fig size/ default: (6, 8)
- fontsize_dict
A dictionary specifying the fontsize of various labels in the plot. default: {‘bigwig_label’: 9, ‘gene_label’: 9, ‘violinplots_xlabel’: 9, ‘title’: 12, ‘bigwig_tick_label’: 5}
- gene_label_offset
The y-offset of the label underneath each gene. default: 3
- arc_rad
The amount of radians the region-to-gene arc should be bend. default: 0.5
- arc_lw
The linewidth of the region-to-gene arcs. default: 1
- cmap_violinplots
A string specifying a matplotlib.cm colormap to use for coloring violinplots. default: ‘Greys’
- violinplots_means_color
A string specifying which color to use to indicate the mean value in violinplots. default: ‘black’
- violinplots_edge_color
A string specifying which color to use for the edge of the violinplots. default: ‘black’
- violoinplots_alpha
The alpha value of the violinplots facecolor. default: 1
- width_ratios_dict
A dict specifying the ratio in vertical direction each part of the plot should use. default: {‘bigwig’: 3, ‘violinplots’: 1}
- height_ratios_dict
A dict specifying the ratio in horizontal direction each part of the plot should use. default: {‘bigwig_violin’: 1, ‘genes’: 0.5, ‘arcs’: 5}
dotplot#
Plot TF expression, motif enrichment and AUC values of target genes and regions in a dotplot.
Export#
Export to loom#
export SCENIC+ object to target genes and target regions loom file.
These files can be visualized in the SCope single cell viewer.
- scenicplus.loom.export_to_loom(scplus_obj: SCENICPLUS, signature_key: str, out_fname: str, eRegulon_metadata_key: str | None = 'eRegulon_metadata', auc_key: str | None = 'eRegulon_AUC', auc_thr_key: str | None = 'eRegulon_AUC_thresholds', keep_direct_and_extended_if_not_direct: bool | None = False, selected_features: List[str] | None = None, selected_cells: List[str] | None = None, cluster_annotation: List[str] = None, tree_structure: Sequence[str] = (), title: str = None, nomenclature: str = 'Unknown')[source]#
Create SCope [Davie et al, 2018] compatible loom files
- Parameters:
scplus_obj (class::SCENICPLUS) – A SCENIC+ object with eRegulons, AUCs, and AUC thresholds computed
signature_key (str) – Whether a ‘Gene_based’ or a ‘Region_based’ file should be produced. Possible values: ‘Gene_based’ or ‘Region_based’
out_fname (str) – Path to output file.
eRegulon_metadata_key (str, optional) – Slot where the eRegulon metadata is stored.
auc_key (str, optional) – Slot where the eRegulon AUC are stored
auc_thr_key (str, optional) – Slot where AUC thresholds are stored
keep_direct_and_extended_if_not_direct (bool, optional) – Keep only direct eregulons and add extended ones only if there is not a direct one.
selected_features (List, optional) – A list with selected genes/region to use.
selected_cells (List, optional) – A list with selected cells to use.
cluster_annotations (List, optional) – A list with variables to use as cluster annotations. By default, those included in the DEGs/DARs dictionary will be used (plus those specificed here)
tree_structure (sequence, optional) – A sequence of strings that defines the category tree structure. Needs to be a sequence of strings with three elements. Default: ()
title (str, optional) – The title for this loom file. If None than the basename of the filename is used as the title. Default: None
nomenclature (str, optional) – The name of the genome. Default: ‘Unknown’
References
Davie, K., Janssens, J., Koldere, D., De Waegeneer, M., Pech, U., Kreft, Ł., … & Aerts, S. (2018). A single-cell transcriptome atlas of the aging Drosophila brain. Cell, 174(4), 982-998. Van de Sande, B., Flerin, C., Davie, K., De Waegeneer, M., Hulselmans, G., Aibar, S., … & Aerts, S. (2020). A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nature Protocols, 15(7), 2247-2276.